Lehrbuch Lyme-Borreliose
23.2 |
Abwehrmechanismen von Bb gegenüber dem humanen Immunsystem |
Das wesentliche medizinische Problem der Lyme-Borreliose ist die Tendenz zur Chronifizierung. Trotz intensiver antibiotischer Behandlung kann die Infektion über Jahre oder gar Jahrzehnte
persistieren. Klinisch handelt es sich um die Lyme-Borreliose im Stadium III (Spätstadium, chronische Lyme-Borreliose) mit einer vielfältigen Symptomatik infolge Multiorgan- oder
Multisystem-Erkrankung.
Der Abwehr des humanen Immunsystems durch Bb liegen zahlreiche Faktoren zu Grunde, die zum Teil auch zugleich die Wirkung der antibiotischen Behandlung abschwächen oder verhindern.
Die wesentlichen Abwehrmechanismen über die Bb gegenüber dem humanen Immunsystem verfügt, sind in Tab. 23.18 aufgelistet.
In einer Übersicht hat P. Kraiczy (103) dargestellt, wie sich Borrelien der Immunabwehr entziehen. Dabei erwähnt er folgende Faktoren:
- Zeckenproteine
- Interaktion mit Komponenten des Komplementsystems
- Interaktion von Borrelien mit Plasminogen
- Interaktion von Borrelien mit extrazellulärer Matrix
- Antigenvariation bei Borrelien
- „Shedding“ von Komplementkomponenten

Immunologische Wirkung von Proteinen des
Zeckenspeichels
Bb bindet mittels OspC ein bestimmtes Zeckenspeichel-Protein (Salp 15). Der Komplex OspC- Salp 15 wird vom humanen Immunsystem nicht erkannt, so dass Bb in dieser sehr frühen Phase nicht
abgetötet wird (1). Die Zeckenspeichelproteine hemmen zudem die Aktivierung und Proliferation von CD4+ T-Zellen durch Bindung an die CD4 Rezeptoren (2, 3), zudem kommt es zur Hemmung von
NK-Zellen, dendritischen Zellen, Makrophagen und Neutrophilen (4, 5, 6, 7). Insbesondere die denditrischen Zellen erfahren im Hinblick auf Proliferation und Funktion erhebliche Einbußen (59,
93-100). Ein im Zeckenspeichel entdecktes 18kDa Protein hemmt die Fähigkeit von OspA und OspC, die B-Zellen-Proliferation zu stimulieren (8) und zwar über einen Zeitraum von etwa 24 Stunden. Auch
das natürliche Immunsystem (Komplementsystem) wird durch Salp 15 von Ixodes scapularis und Ixodes ricinus gehemmt (52).
Usurpation des wirtseigenen Plasminogen-Aktivator-Systems
OspC bildet einen plasminogenen Rezeptor. Durch Bindung von Plasminogen an diesen Rezeptor erhält Bb die Fähigkeit, die Wirkung von Plasminen zu nutzen. Plasmin löst enzymatisch Fibrin und große
Glucoproteine und öffnet Bb damit den Weg, aus dem Kreislaufsystem in die extrazelluläre Matrix (ECM) zu gelangen. Dort entkommt Bb den Immunzellen und Antikörpern, die sich in Lymphe und Blut
befinden (9). Entsprechend wurde tierexperimentell nachgewiesen, dass die Invasions-Fähigkeit von der genomischen Variation des OspC abhängt (10). Der Plasmin-assoziierte Mechanismus dient
möglicherweise auch dazu, die Blut-Hirnschranke zu überwinden und eine Invasion des zentralen Nervensystems zu ermöglichen (11).
Einfluss von Bb auf das Komplementsystem
Das Komplementsystem kann auf 3 verschiedenen Wegen aktiviert werden: klassischer Aktivierungsweg, alternativer Aktivierungsweg, Lektin-Aktivierungsweg. Der klassische Aktivierungsweg wird durch
Antikörper induziert, welche sich an die für sie spezifischen Antigene gebunden haben. Dabei wird die molekulare Struktur der Antikörper verändert, so dass Bindung an die erste Stufe des
Komplementsystems (C1) ermöglicht wird. Auch die direkte Bindung von C1 an die Bakterienoberfläche kann die Komplementkaskade aktivieren (12). Der alternative Weg wird getriggert, wenn das
Komplement-Protein C3b eine Verbindung mit der Oberfläche eines Pathogens eingeht, sofern der Faktor H sich nicht auf der Oberfläche befindet und dadurch die Bildung des Erreger-vernichtenden MAC
verhindert (11). Der Lektin-Weg wird getriggert durch Mannin-bindendes Lektin, das sich an glukolisierte Moleküle auf der Oberfläche des Pathogenes bindet (13). Über den Lektin-Weg werden die
Lektin-assoziierte Serin-Proteasen aktiviert, die den klassischen und alternativen Weg des Komplementsystems verstärken.
Das Komplementsystem besteht aus den Komponenten C1-C9 und deren Subkomponenten. Anhaftung der Mikrobe (Bb) an C1, Aktivierung und Verstärkung des Komplementsystems bei C2-C4, Bildung von terminalem Komplement-Komplex (TCC) (syn. „Membrane Attack Complex“ (MAC)) bei C5b-C9 (Abb. 23.9).
Generell aktiviert Bb das Komplementsystem über den alternativen oder klassischen Weg (53). Wahrscheinlich erfolgt die Aktivierung sogar nur über den klassischen Weg (54).
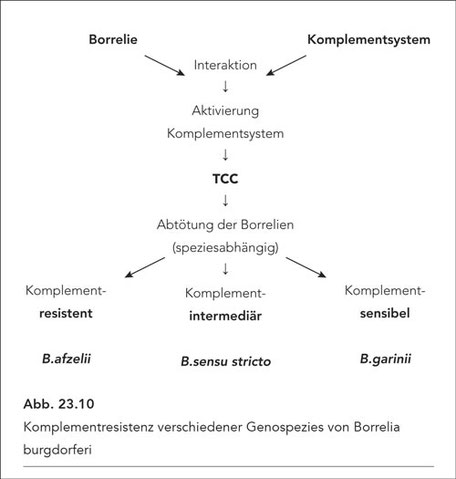
Die Reaktion des Komplementsystems auf Kontakt mit Bb ist in Form einer Übersicht in Abb. 23.10 dargestellt. Es zeigt sich, dass verschiedene Genospezies gegenüber dem Komplementsystem
unterschiedlich sensibel sind: B. afzelii ist resistent, B. sensu stricto ist begrenzt resistent (intermediär), B. garinii ist Komplement-sensibel (53).
Das Komplementsystem wird durch Regulatorproteine gehemmt, die sich auf der Oberfläche von Bb anlagern. Die wichtigsten Regulatorproteine sind Faktor H und FHL-1/Reconectin. Faktor H inaktiviert
Kaskadenstufen durch Inhibierung der Konvertasen bei C3 und C5 (55, 56, 57, 58). Borrelien sind in der Lage, FHL-1/Reconectin und Faktor H aus dem Serum des Wirtes (Mensch) zu absorbieren. Eine
solche Bindung von Regulatoren auf der Bakterienoberfläche wurde bei der Komplement-resistenten Borrelia afzelii nachgewiesen, nicht jedoch bei der Komplement-sensiblen Borrelia garinii (60, 61,
62, 67). Auch ist Bb befähigt, selbst Regulator-Proteine zu produzieren (53).
Die Anhäufung von Regulatorprotein auf der Bakterienoberfläche ist bei Komplement-resistenten Subspezies (B. afzelii) sehr viel größer als bei Intermediär-sensiblen. Bei Komplement-sensiblen (B.
garinii) sind Proteinregulatoren auf der Oberfläche überhaupt nicht nachweisbar. Komplement-resistente Bb sind also schwache Komplement-Aktivatoren, sensible Borrelien starke Aktivatoren (53).
Bei den Komplement-sensitiven Borrelien (B. garinii) führt die Komplement-Aktivierung zu einer massiven Ablagerung von C3b auf der Borrelien-Oberfläche. Die resultierende Aktivierung der
Komplementkaskade führt zur Produktion von TCC, das in die Bakterienmembran eindringt mit der Folge der Bakteriolyse (53).
Die Bindung der Komplement-hemmenden Regulatorproteine erfolgt durch CRASPs („complement regulator aquiring surface proteins“). Eine solche Bindung wurde insbesondere für FHL-1/Reconectin und
Faktor H auf B. afzelii und B. sensu stricto nachgewiesen, nicht jedoch bei B. garinii (53).

Ein besonderer Angriffspunkt der Regulatorproteine ist die Kaskadenstufe C3b. Die Regulatorproteine reduzieren die Bildung von C3b auf der Bb-Oberfläche und reduzieren zusätzlich die Aktivität der involvierten C3-Konvertase. Inaktiviertes C3b (iC3b) wurde experimentell nachgewiesen. Ein solcher Vorgang wird bei Komplement-resistenten, nicht jedoch bei Komplement-sensiblen Borrelien gefunden (53, 63).
Faktor H bindet auch an OspE oder OspE Varianten. Daher wird OspE als das Molekül aufgefasst, das es Bb ermöglicht, dem Komplementsystem zu entkommen (16). Die Expression von OspE wird
getriggert, wenn Signale einer Blutmahlzeit den Mitteldarm erreichen, in dem sich die Borrelien üblicherweise aufhalten (17).
Die Komplementresistenz bzw. die Komplementsensitivität findet ihr Korrelat in der Menge von TCC (terminaler Komplementkomplex, Synonym MAC, „Membrane Attack Complex“). Bei Komplement-resistenten
Bakterien findet sich eine TCC-Ablagerung nur in 5-20%, bei Komplement-sensiblen bei 60% der Bakterien (15, 64, 65, 66, 67, 68).
Auch die Anlagerung von C3b ist unterschiedlich. Bei Komplement-resistenten beträgt sie 5-20% der Bakterien, bei Intermediär-resistenten 25% und bei Sensitiven 20-60% oder in etwa gleicher
Größenordnung nach den Befunden anderer Autoren (53, 69). Bei den Komplement-resistenten Borrelien (Ba) wird also der Großteil der Borrelien vom Komplement nicht abgetötet, im Gegensatz zu der
Komplement-sensiblen B. garinii, bei der nur ein geringer Anteil der Vernichtung durch Komplement entgeht.
B. garinii (Komplement-sensitiv) kann in vitro über Monate im Komplement-haltigen Blasengewebe von Mäusen oder Nervengewebe von humanen Primaten überleben (70). Die Erkenntnisse in vitro sind
also nicht ohne weiteres auf in vivo übertragbar.
Möglicherweise sind bei der Komplementresistenz weitere Faktoren beteiligt, nämlich: Oberlächenantigene A, B, C oder D, Dekorin-bindende Proteine (DbpA) und sonstige Oberlächenproteine (60, 61,
71, 72). Auch existieren weitere CRASP (CRASP3, CRASP4 und CRASP5) (73). Auf Komplement-sensitiven Borrelien (B. garinii) sind CRASP nicht oder in nur unbedeutender Menge vorhanden (60, 74).
Außer CRASP können auch andere Oberflächenproteine Komplementregulatoren binden (75).
Bei Komplement-resistenten Bb wurde das Glykoprotein CD59 nachgewiesen, das die Fähigkeit besitzt, die Kaskadenstufen C8 und C9 zu binden und damit die Bildung von TCC (MAC) zu verhindern
(76).
Ein weiterer Schutzmechanismus gegen das Komplementsystem ist die Bildung einer schleimartigen Struktur, die Bb umgibt und die eine Insertion von TCC verhindert. Komplement-resistente Bb können
schleimartige Strukturen mit dem darauf abgelagerten TCC abstreifen („Shedding“), ein Vorgang, der zur Komplementresistenz beiträgt. Komplement-sensible Borrelien haben eine derartige
Schleimschicht nicht (53). In Europa ist Borrelia afzelii die am häufigsten vorkommende Subspezies, d.h. es muss entsprechend oft mit Komplement-resistenten Borrelien gerechnet werden (77).
Diversifizierung von Oberflächenproteinen (Osp)
Die Diversifizierung von Oberflächenantigenen (Epitopen) ist ein üblicher Mechanismus von Krankheitserregern, sich dem Immunsystem des Wirtsorganismus zu entziehen (19). Bei Bb erfolgt die
Diversifizierung im Wesentlichen durch das VlsE-System (20). Genetisch ist der Mechanismus lokalisiert im linearen Plasmid lp28-1 (20, 21). Die Diversifizierung basiert auf einem Kassettensystem
(22). Die zentrale VlsE-Kassette steht in ständigem Kontakt mit 15 schweigenden Kassetten, die immer wieder verschiedene Aminosäuren bereitstellen und so die Diversifizierung der Epitopen
ermöglichen (22).
Die Diversifizierung wird im Wesentlichen durch die Branch Migrase ruvA vermittelt (23). Bei Mutation von ruvA und ruvB ist die Infektion von Bb reduziert und die antibiotische Behandlung ist
effektiver (23).
Bezüglich der biochemischen Einzelheiten im Zusammenhang mit der VlsE-Diversifizierung sei auf das Kap. 23.5 verwiesen.
Die Oberflächenantigene (Epitope, OSP) sind auf der äußeren Membran von Bb angeordnet. Bei Kontakt mit dem erworbenen Immunsystem produzieren spezifische B-Zellen unter Mitwirkung anderer
immunologischer Faktoren Antikörper. Die Bindung der Antikörper an die Antigene leitet die Bakteriolyse ein (vgl. Abb. 23.11).
Das Prinzip der Diversifizierung ist in Abb. 23.12 schematisch dargestellt. Das Antigen Ag1 induziert über das erworbene Immunsystem die Bildung des entsprechenden Antikörpers AK1. Noch bevor die
Antikörperbildung abgeschlossen ist, hat sich das Antigen Ag1 durch Diversifizierung in das anders strukturierte Antigen Ag2 umgewandelt. AK1 ist mit Ag2 nicht kompatibel und somit immunologisch
unwirksam.


Motilität von Bb
Motilität und chemotaktische Funktionen sind bei Bb sehr viel ausgeprägter als bei Bakterien im Allgemeinen. Bb ist in der Lage, sich in Flüssigkeiten (Blut, Lymphe, Liquor) und in
viskoelastischem Gel (z.B. ECM oder andere Bindegewebsstrukturen) fortzubewegen (25). Bb dockt an Epithelzellen an, meistens im Bereich der Endothelzellen-Verbindungen (26). Bb ist in der Lage,
die Gefäßwand zu passieren und in die ECM einzudringen. Ein solcher Vorgang dauert durchschnittlich knapp 11 Minuten. Dabei werden Penetrationsgeschwindigkeiten von gut 0,5 mm/min erreicht (26).
Im Vergleich zu anderen Mikroben ist eine derartige Bewegungsgeschwindigkeit ungewöhnlich hoch; sie ermöglicht Bb die Infiltration des Gewebes und die Vermeidung des Immunsystems. Die hohe
Motilität ist somit wesentlich für die Persistenz der Infektion.
Chemotaxis und Aufenthalt in schwer erreichbaren Kompartimenten
Bb verfügt über das höchst entwickelte Chemotaxie-bezogene Gen in der Gruppe der Eubakterien (27). Bb benutzt Chemorezeptoren an den beiden Zellpolen, um auf chemotaktischem Weg spezifische
Wirtszellen oder Gewebskompartimente zu erreichen (28, 29). Ohne chemotaktische Eigenschaften verliert Bb seine Infektiosität (30). Bb benutzt Adhäsine zur Anbindung an viele Moleküle,
insbesondere Glykosaminoglycane (GAG), Decorin und Fibronektin (31, 32, 33).
Die extrazelluläre Matrix (ECM) ist eine netzartige Struktur aus interstitiellem Gewebe, Basalmembran, Kollagen und Polysaccharid-Gelen. Die ECM ist der bevorzugte Aufenthaltsort von Bb. Die meist untersuchten Adhäsine von Bb sind die Decorin-bindenden Proteine (Dbp). Antikörper gegen DbpA führten im Tierexperiment zur Rückbildung von entzündlichen Veränderungen in Gelenken und Herz (34). Die Chemo-Attraktion zwischen Bb und Kollagen ist so groß, dass eine Invasion der Collagennetze auch in Abwesenheit von Decorin erfolgt (35).
Der Aufenthalt in der ECM verhilft Bb offensichtlich dem humanen Immunsystem zu entkommen. Dies zeigt sich insbesondere auch in Tierexperimenten, in denen trotz ausgeprägter Reaktion des
natürlichen und des erworbenen Immunsystems die Infektion persistierte, insbesondere in Sehnen und Bändern (36, 37). Die Literatur zeigt, dass Bb eine starke Affinität zu den Molekülen der ECM
hat, in der das Immunsystem weniger wirksam ist, wahrscheinlich eine weitere wesentliche Bedingung für die Persistenz der Infektion.
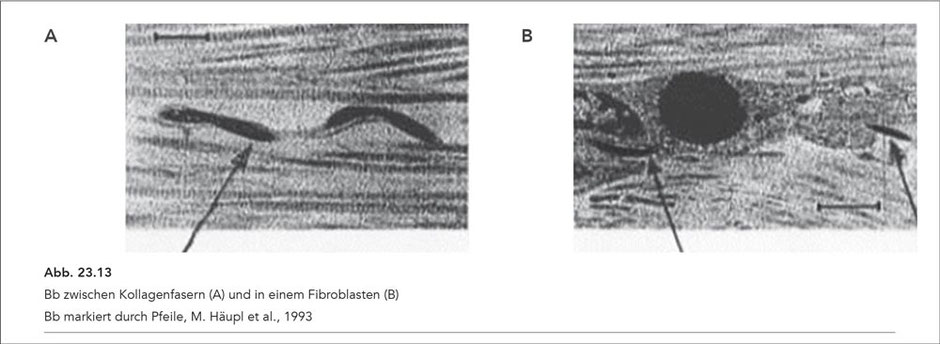
Der Aufenthalt von Bb im kollagenen Gewebe und als ergänzende Darstellung auch in einem Fibroblasten zeigt die Abb. 23.13.
Bb ist in Fibroblasten vor Antibiotika geschützt (78). Auch der Aufenthalt in Synovialiszellen ist experimentell nachgewiesen (79). In vitro wurde dasselbe Phänomen an humanen Synovialiszellen
gefunden, in denen Bb selbst nach neuntägiger Ceftriaxon-Exposition überlebte (80). Auch das Eindringen von Bb in Endothelzellen von Umbilikalvenen wurde experimentell nachgewiesen (81).
Gewebsnischen, das heißt z.B. Sehnen, Bindegewebe, Knorpel sind für das Immunsystem nicht oder nur schlecht erreichbar (82). Bb war zwischen Kollagenfasern und in Fibroblasten nachweisbar trotz
vorausgegangener Behandlung der Patienten mit Ceftriaxon (83).
Bei Inkubation von Borrelia burgdorferi (Bb) zusammen mit humanen umbilikalen Venen-Endothelzellen waren nach 24 Stunden 10-25% der Bb intrazellulär lokalisiert. Actin-enthaltende Mikrofilamente
waren für die intrazelluläre Lokalisation erforderlich; dies zeigt, dass die Wirtszellen an der Internalisierung partizipieren. Der Endothelrezeptor für Bb ist konstitutionell exprimiert. Die
Internalisierung ist nicht von Adhäsionsmolekülen abhängig. Der Aufenthalt von Bb in den Endothelzellen legt nahe, dass die intrazelluläre Lokalisation dem Erreger ermöglicht, sich dem
Immunsystem zu entziehen und so zur Persistenz der Infektion im Spätstadium beiträgt (81).
Bildung von Schutzfilmen
Die Bildung einer Schleimschicht mit aufgelagertem TCC (terminalem Komplementkomplex (syn. „Membrane Attack Complex“ (MAC)) wurde oben im Zusammenhang mit dem Komplementsystem bereits
dargestellt. Diese schleimige Schutzschicht mit TCC kann abgestreift werden („Shedding“).
Schutzfilme auf Bb können auch aus Dekorin oder Fibronektin bestehen. Das Glykoprotein Dekorin befindet sich insbesondere in Strukturen, die reich an extrazellulärer Matrix sind (Gelenke, Haut,
Knorpel, Sehnen). Das Dekorin bindet an Dekorin-bindende Antigene (DbpA), die sich auf der Oberfläche von Bb befinden. Diese Dekorinschicht stellt einen Abwehrmechanismus gegenüber dem
wirtseigenen Immunsystem dar (84).
Auch Fibronektin als ein weiteres Hauptgylokoprotein der extrazellulären Matrix (ECM) wird von Bb über das Oberflächenantigen BBK32 (p47) gebunden. Das Fibronektin dient Bb, sich an
Zellstrukturen zu fixieren und begünstigt damit die Kolonisation des Gewebes (77). Die Begünstigung der Kolonisation könnte eine erste Phase zur Bildung Biofilm-ähnlicher Strukturen darstellen
(vgl. Abb. 23.14 und 23.15).
Biofilmbildung bei Bb
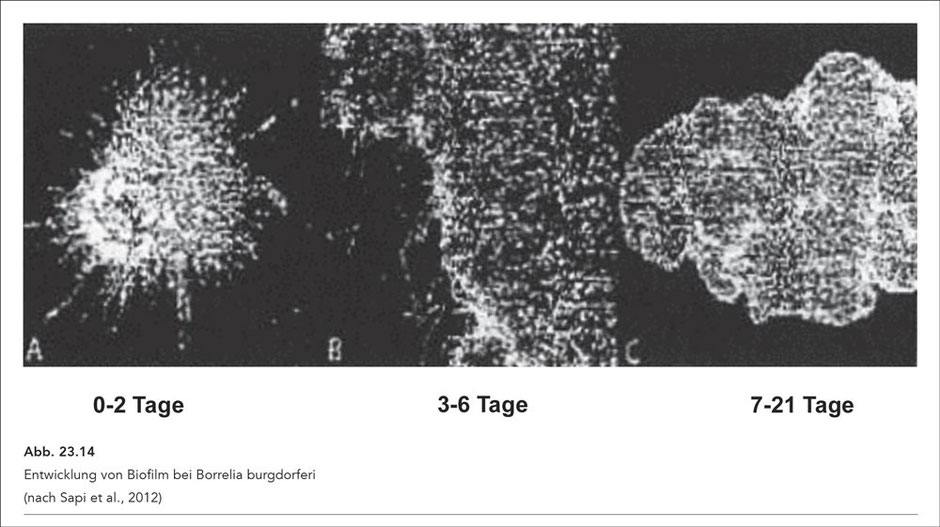
Biofilme sind ein wesentlicher Grund für eine persistierende Infektion (38, 39, 40, 41, 42). Bei Bb wurde die Bildung von Biofilm erstmalig durch Sapi et al. nachgewiesen (43). Bei der chemischen
Analyse der extrazellulären Substrate fanden sich Polysaccharide einschließlich Alginate, die eine gummiartige Schicht um Bb bildeten und gewissermaßen eine Quelle für Ernährung und Hydration
darstellen. Auch wurde extrazelluläre DNA in den Aggregaten nachgewiesen, die für die Bildung von Biofilmen erforderlich sind (44). Sapi und Mitarbeiter folgerten, dass Biofilme auch in vivo im
Zusammenhang mit Bb vorkommen und einen Schutz gegen den Umgebungsstress im Wirtsorganismus bieten.
Dieselbe Arbeitsgruppe zeigt in einer nachfolgenden Studie die Biofilmbildung in vivo und zwar im Biopsaten von Borrelien-Lymphozytomen. Bb-Aggregate exprimierten Biofilmmarker in Form von
protektiven Schichten aus verschiedenen Mucopolysachariden, insbesondere von Alginaten. Überdies bestand Hinweis auf die typischen Merkmale von Biofilmen, nämlich Kanäle und Protrusionen an der
Oberfläche (108).
Die Biofilm-Entwicklung und die von einem Biofilm umgebenen Bb-Haufen sind in Abb. 23.14 dargestellt.
Die Anhäufung von Bb (Kolonie-artige Massen) im Gehirn wurde von Miklossy et al., 2004 nachgewiesen (vgl. Abb. 23.15). Bb verfügt offensichtlich über die Fähigkeit sich in großer Zahl
anzusammeln, gewissermaßen der erste Schritt auf dem Weg zur Bildung Biofilm umgebener Aggregate (50). In vitro waren solche Koloniebildungen bereits durch Kurtti et al., 1987 nachgewiesen worden
(104).

Horizontaler Gentransfer
Bakterien können Gene vertikal auf Tochterzellen übertragen oder durch horizontalen Gentransfer und zwar mittels Transformation, Konjugation oder Transduktion. Der horizontale Gentransfer
überträgt und verbreitet bei Bakterien, auch bei Bb, die Fähigkeit, sich gegenüber ungünstigen Einflüssen zu schützen. Dies betrifft z. B. die Abwehr des Komplementsystems oder die Bildung von
Biofilm (45, 46).
Beim Gentransfer können auch Bakteriophagen an der Übertragung von DNA-Anteilen (Kassetten) von einem Stamm auf einen anderen Stamm von Bb beteiligt sein; zugleich wurde experimentell
nachgewiesen, dass der horizontale Gentransfer die Übertragung einer Antibiotikaresistenz von einem Stamm zum anderen ermöglicht (105, 106).
Atypische Formen
Unter ungünstigen Bedingungen ändert Bb seine Form. Dabei können ganz unterschiedliche Strukturen entstehen, sogenannte Pleomorphie (47, 48). Andere Experimente zeigten, dass bei Inokulation von
Bb in Liquor innerhalb von 1-24 Stunden zystische Formen entstanden; nach Rückführung in ein adäquates Medium (BSK) entstanden aus diesen zystischen Formen wieder normale, mobile Spirochäten
innerhalb von 9-17 Tagen (85, 86). Die zystischen Formen sind für das Immunsystem und für Antibiotika, ausgenommen Nitroimidazole und Tygecyclin, nicht angreifbar. Zystische Formen bleiben
infektiös (87).
Die Pleomorphie bei Bb umfasst auch zahlreiche weitere Formanomalien: atypische Zysten, eingerollte Spirochäten, schlangenartige Formen, Ringformen, globuläre und sphärische Formen sowie granuläres Material (Abb. 23.16 und 23.17).
Degenerierte Formen von Bb wurden auch in den fibrosklerotischen Anteilen eines Morphaea, also in vivo, nachgewiesen (102).
Auch bei diesen Formen ist die Einwirkung des Immunsystems und die Wirkung von Antibiotika mehr oder weniger eingeschränkt oder aufgehoben.
Atypische Formen wurden auch in vivo sowie post mortem (im Gehirn) nachgewiesen (49). Zudem entdeckte Miklossy et al. (49), dass Bb Beta-Amyloid-Ablagerungen und andere Veränderungen induziert,
wie sie bei der Alzheimer Krankheit beobachtet werden.
Die Entwicklung atypischer Bakterienformen ist eine Änderung der Morphologie unter Stressbedingungen (Hungerreaktion), d.h. Entzug von Serum, Fettsäuren, Sauerstoff. Auch erhöhte Temperaturen,
saures Milieu und insbesondere Antibiotika wurden als Ursachen experimentell nachgewiesen (88, 89).
Zysten haben einen minimalen Stoffwechsel; daraus lässt sich folgern, dass Zysten keine Degenerationsprodukte sind, sondern Überlebensformen darstellen (77).
Zystenbildung in vitro unter dem Einfluss von Antibiotika ist experimentell belegt (90 91). Unter der Einwirkung von Antibiotika wird auch die Motilität von Bb eingeschränkt (91), jedoch zeigte
sich in diesen Experimenten, dass trotz der antibiotischen Belastung intakte Spirochäten noch nach 4 Tagen nachweisbar waren.
Atypische Formen wurden auch im Gewebe und im Blut nachgewiesen (92). In den Experimenten zeigte sich unter dem Einfluss von Antibiotika eine verminderte Motilität, verlangsamtes Wachstum,
herabgesetzte Vermehrungsfähigkeit, Bildung von Kolonien, kein Ansprechen auf Oberflächen-wirksame Antibiotika. Die zystischen Formen entstehen durch Ausstülpung der Zellwand; folglich befinden
sich auf der Oberfläche der Zysten keine Oberflächenantigene. Die Folge ist Unempfindlichkeit gegenüber dem Immunsystem und den Oberflächen-wirksamen Antibiotika. Die zystischen Formen können
folglich persistieren; unter günstigen Bedingungen entwickeln sich wieder normale spirochätale Borrelien (92).
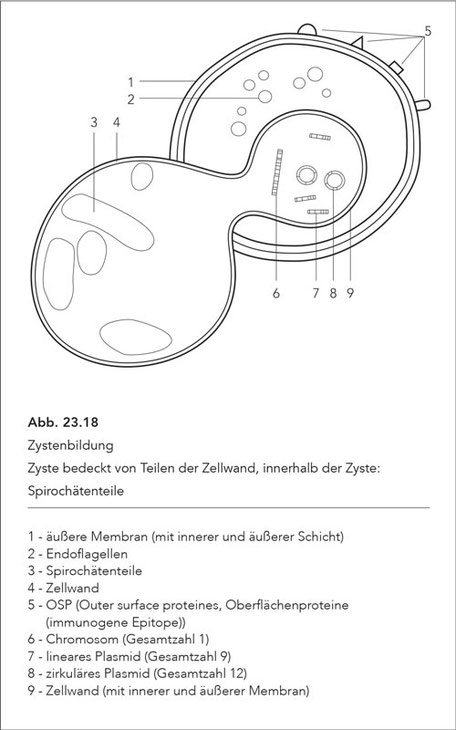
Die Bildung von Zysten ist schematisch in Abb. 23.18 dargestellt. Bei dem Vorgang der Zystenbildung stülpt sich die Zellwand nach außen vor, die Zysten können sich ablösen („Blebs“) und enthalten
Anteile von Bb (Abb. 23.19). Die Oberfläche der Zysten besteht aus Teilen der Zellwand und trägt somit keine Epitope, die (normalerweise) auf der äußeren Zellmembran lokalisiert sind. Das Fehlen
der Epitope macht die Zysten für das erworbene Immunsystem unangreifbar.
Der Vollständigkeit halber sei angemerkt, dass bei der morphologischen Veränderung von Bb und der damit verbundenen Abwehr des Immunsystems zunehmend die sogenannten „Round bodies“ in den Fokus
treten (vgl. Kap. 23.4). Wie solche „Round bodies“ mit der aktuell diskutierten Struktur sich dem Immunsystem erwehren oder entziehen ist wissenschaftlich ungeklärt.



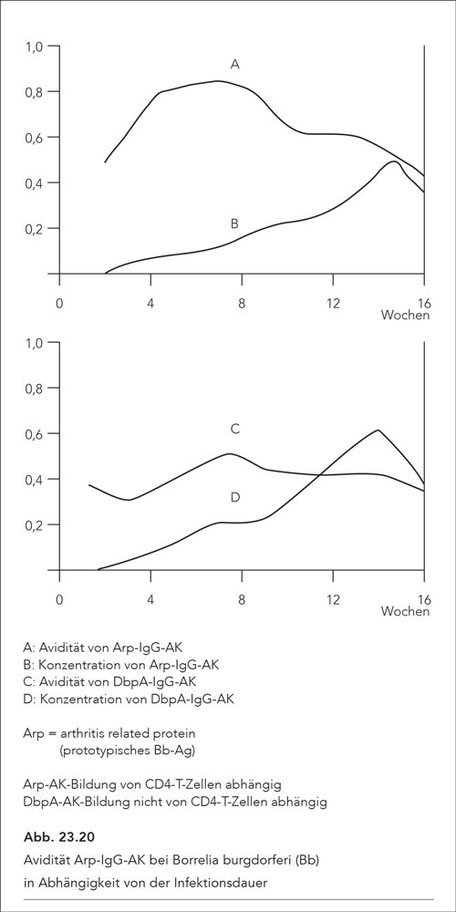
Verminderung der Avidität von Bb-AK
Die Erkenntnis, dass Bb möglicherweise die Avidität (Bindungshaft) von Bb-AK an die Antigene auf der Borrelien-Oberfläche vermindert, geht auf die Studie von Elsner et al., 2015 zurück.
CD4-T-Zellen sind entscheidend für die B-Zellen-vermittelte Immunität, d.h. für die Bildung von Antikörpern, langlebigen Plasmazellen und Gedächtnis-B-Zellen.
Experimentell wurde nachgewiesen, dass Bb offensichtlich zu einer mangelhaften T-abhängigen-B-Zell-Antwort führt, es kommt im Verlauf der Infektion nicht zu einer über Monate anhaltenden Bildung
von Plasmazellen und Gedächtnis-B-Zellen. – Auch werden zwar ständig IgM-AK während der chronischen Lyme-Borreliose produziert, jedoch ohne Umwandlung in IgG-AK.
In der Studie von Elsner et al., 2015 wurde im Tierexperiment gezeigt, dass eine Bb-Infektion der Produktion von protektiven, hochaffinen und langlebigen AK entgegenwirkt und vielmehr eine rasche
ausgeprägte, jedoch kurzlebige Bildung von AK begünstigt mit begrenzter Effizienz. Die Abnahme der Avidität beginnt etwa 6 Wochen nach Infektionsbeginn, die Rückbildung germinaler Zentren bereits
nach 3-4 Wochen.
Im Verlauf einer späten Bb-Infektion führt die Immunantwort zur Bildung langlebiger AK und Gedächtnis-B-Zellen. Allerdings hat zu diesem Zeitpunkt bereits eine Dissemination der Bakterien
stattgefunden, so dass die Immunantwort den Erreger nicht mehr voll erreicht. Das Versagen der germinalen Zentren erklärt die häufige Neigung zur Reinfektion durch Bb.
Die nach Bb-Infektion zunehmende Antikörperbildung bei allerdings gleichzeitiger Abnahme der Avidität ist in Abb. 23.20 dargestellt.
Bezüglich der Einzelheiten sei auf die Epikrise (in deutscher Übersetzung) der Publikation von Elsner et al., 2015, Kap. 23.6 verwiesen.
Zusammenfassung
Bb tritt rasch aus dem Gefäßsystem in die ECM über. Es besteht der Hinweis, dass Bb Biofilme bildet.
In Tierversuchen wurde nachgewiesen, dass LB eine schwer zu beeinflussende, chronisch-rezidivierende Infektion ist.
Weder die antibiotische Behandlung nach Standard noch eine aggressivere Antibiose sind in der Lage, Bb in der Maus zu beseitigen. Vielmehr kommt unter Antibiose die Zellteilung von Bb zum
Erliegen oder wird deutlich verlangsamt, jedoch erhalten auch diese vitalitätsgeminderten Bb offensichtlich ihre Infektiosität.
Unter der Federführung der IDSA (Infectious Diseases Society of America) wird in den nationalen und internationalen Leitlinien eine Behandlung nach Standard empfohlen, also in aller Regel eine
antibiotische Monotherapie für höchstens 4 Wochen. Diese Behandlung nach Standard wird als von der IDSA und vielen sonstigen Fachgesellschaften adäquat betrachtet. Tatsächlich zeigen jedoch
zahlreiche Studien, dass eine solche Behandlung nach Standard die Infektion nicht beseitigt, vielmehr persistiert die Infektion. Wissenschaftlich fehlen Grundlagen, die einen Weg zeigen, um die
persistierende Infektion angemessen zu behandeln.
- Literaturverzeichnis
-
- Ramamoorthi N, Narasimhan S, Pal U, Bao F, Yang XF, Fish D, Anguita J, Norgard MV, Kantor FS, Anderson JF, Koski RA, Fikrig E. The Lyme disease agent exploits a tick protein to infect the mammalian host. Nature. 2005; 28;436(7050):573-7.
- Anguita J, Ramamoorthi N, Hovius JW, Das S, Thomas V, Persinski R, Conze D, Askenase PW, Rincón M, Kantor FS, Fikrig E. Salp15, an ixodes scapularis salivary protein, inhibits CD4(+) T cell activation. Immunity. 2002; 16(6):849-859.
- Juncadella IJ, Garg R, Ananthnarayanan SK, Yengo CM, Anguita J. T-cell signaling pathways inhibited by the tick saliva immunosuppressor, Salp15. FEMS Immunol Med Microbiol. 2007; 49(3):433-438.
- Kubes M, Kocáková P, Slovák M, Sláviková M, Fuchsberger N, Nuttal PA. Heterogeneity in the effect of different ixodid tick species on human natural killer cell activity. Parasite Immunol. 2002; 24(1):23-28.
- Hovius JW, de Jong MA, den Dunnen J, Litjens M, Fikrig E, van der Poll T, Gringhuis SI, Geijtenbeek TB. Salp15 binding to DC-SIGN inhibits cytokine expression by impairing both nucleosome remodeling and mRNA stabilization. PLoS Pathog. 2008; 4(2):e31.
- Gwakisa P, Yoshihara K, Long To T, Gotoh H, Amano F, Momotani E. Salivary gland extract of Rhipicephalus appendiculatus ticks inhibits in vitro transcription and secretion of cytokines and production of nitric oxide by LPS-stimulated JA-4 cells. Vet Parasitol. 2001; 99(1):53-61.
- Montgomery RR, Lusitani D, De Boisfleury Chevance A, Malawista SE. Tick saliva reduces adherence and area of human neutrophils. Infect Immun. 2004; 72(5):2989-2994.
- Hannier S, Liversidge J, Sternberg JM, Bowman AS. Characterization of the B-cell inhibitory protein factor in Ixodes ricinus tick saliva: a potential role in enhanced Borrelia burgdorferi transmission. Immunology. 2004; 113(3):401-408.
- Önder Ö, Humphrey PT, McOmber B, Korobova F, Francella N, Greenbaum DC, Brisson D. OspC is potent plasminogen receptor on surface of Borrelia burgdorferi. J Biol Chem. 2012; 287(20):16860-16868.
- Lagal V, Portnoï D, Faure G, Postic D, Baranton G. Borrelia burgdorferi sensu stricto invasiveness is correlated with OspC-plasminogen affinity. Microbes Infect. 2006; 8(3):645-652.
- Grab DJ, Perides G, Dumler JS, Kim KJ, Park J, Kim YV, Nikolskaia O, Choi KS, Stins MF, Kim KS. Borrelia burgdorferi, host-derived proteases, and the blood-brain barrier. Infect Immun. 2005; 73(2):1014-1022.
- Daha NA, Banda NK, Roos A, Beurskens FJ, Bakker JM, Daha MR, Trouw LA. Complement activation by (auto-) antibodies. Mol Immunol. 2011; 48(14):1656-1665.
- Thiel S, Gadjeva M. Humoral pattern recognition molecules: mannin-binding lectin and ficolins. Adv Exp Med Biol. 2009; 653:58-73.
- Schuijt TJ, Hovius JW, van Burgel ND, Ramamoorthi N, Fikrig E, van Dam AP. The tick salivary protein Salp15 inhibits the killing of serum-sensitive Borrelia burgdorferi sensu lato isolates. Infect Immun. 2008; 76(7):2888-2894.
- Alitalo A, Meri T, Rämö L, Jokiranta TS, Heikkilä T, Seppälä IJ, Oksi J, Viljanen M, Meri S. Complement evasion by Borrelia burgdorferi: serum-resistant strains promote C3b inactivation. Infect Immun. 2001; 69(6):3865-3891.
- Alitalo A, Meri T, Lankinen H, Seppälä I, Lahdenne P, Hefty PS, Akins D, Meri S. Complement inhibitor factor H binding to Lyme disease spirochetes is mediated by inducible expression of multiple plasmid-encoded outer surface protein E paralogs. J Immunol. 2002;1 69(7):3847-3853.
- Hefty PS, Jolliff SE, Caimano MJ, Wikel SK, Akins DR. Changes in temporal and spatial patterns of outer surface lipoprotein expression generate population heterogeneity and antigenic diversity in the Lyme disease spirochete, Borrelia burgdorferi. Infect Immun. 2002; 70(7):3468-3478.
- Kraiczy P, Skerka C, Kirschfink M, Brade V, Zipfel PF. Immune evasion of Borrelia burgdorferi by acquisition of human complement regulators FHL-1/reconectin and Faktor H. Eur J Immunol. 2001; 31:1674-1584.
- Vink C, Rudenko G, Seifert HS. Microbial antigenic variation mediated by homologous DNA recombination. FEMS Microbiol Rev. 2011; 1574-6976.
- Zhang JR, Hardham JM, Barbour AG, Norris SJ. Antigenic variation in Lyme disease borreliae by promiscuous recombination of VMP-like sequence cassettes. Cell. 1997; 89(2):275-285.
- Kawabata H, Myouga F, Inagaki Y, Murai N, Watanabe H. Genetic and immunological analyses of VIs (VMP-like sequences) of Borrelia burgdorferi. Microb Pathog. 1998; 24(3):155-166.
- Norris SJ. Antigenic variation with a twist – the Borrelia story. Mol Microbiol. 2006; 60(6):1319-1322.
- Dresser AR, Hardy PO, Chaconas G. Investigation of the genes involved in antigenic switching at the vlsE locus in Borrelia burgdorferi: an essential role for the ruvAB branch migrase. PLoS Pathog. 2009; 5(12):e1000680.
- Lin T, Gao L, Edmonson DG, Jacobs MB, Philipp MT, Norris SJ. Central role of the Holliday junction helicase RuvAB in vIsE recombination and infectivity in Borrelia burgdorferi. PLoS Pathog. 2009; 5(12):e1000679.
- Kimsey RB und Spielman A. Motility of Lyme disease spirochetes in fluids as viscous as the extracellular matrix. J Infect Dis. 1990; 162(5):1205-1208.
- Moriarty TJ, Norman UM, Colarusso P, Bankhead T, Kubes P, Chaconas G. Real-time high resolution 3D imaging of the lyme disease spirochete adhering to and escaping from the vasculature of a living host. PLoS Pathog. 2008; 4(6):e1000090.
- Fraser CM, Casjens S, Huang WM, Sutton GG, Clayton R, Lathigra R, White O, Ketchum KA, Dodson R, Hickey EK, Gwinn M, Dougherty B, Tomb JF, Fleischmann RD, Richardson D, Peterson J, Kerlavage AR, Quackenbush J, Salzberg S, Hanson M, van Vugt R, Palmer N, Adams MD, Gocayne J, Weidman J, Utterback T, Watthey L, McDonald L, Artiach P, Bowman C, Garland S, Fuji C, Cotton MD, Horst K, Roberts K, Hatch B, Smith HO, Venter JC. Genomic sequence of a Lyme disease spirochete, Borrelia burgdorferi. Nature. 1997; 390(6660):580-586.
- Xu H, Raddi G, Liu J, Charon NW, Li C. Chemoreceptors and flagellar motors are subterminally located in close proximity at the two cell poles in spirochetes. J Bacteriol. 2011; 193(10):2652-2656.
- Zhang K, Liu J, Tu Y, Xu H, Charon NW, Li C. Two CheW coupling proteins are essential in a chemosensory pathway of Borrelia burgdorferi. Mol Microbiol. 2012; 85(4):782-794.
- Sze CS, Zhang K, Kariu T, Pal U, Li C. Borrelia burgdorferi needs chemotaxis to establish infection in mammals and to accomplish its enzootic cycle. Infect Immun. 2012; 80(7):2485-2492.
- Parveen N und Leong JM. Identification of a candidate glycosaminoglycan-binding adhesin of the Lyme disease spirochete Borrelia burgdorferi. Mol Microbiol. 1998; 35(5):1220-1234.
- Guo BP, Brown EL, Dorward DW, Rosenberg LC, Höök M. Decorin-binding adhesins from Borrelia burgdorferi. Mol Microbiol. 1998; 30(4):711–723.
- Probert WS und Johnson BJ. Identification of a 47 kDa fibrinonectin-binding protein expressed by Borrelia burgdorferi isolate B31. Mol Microbiol. 1998; 30(5):1003-1015.
- Barthold SW, Hodzic E, Tunev S, Feng S. Antibody-mediated disease remission in the mouse model of lyme borreliosis. Infect Immun. 2006; 74(8):4817-4825.
- Zambrano MC, Beklemisheva AV, Bryksin AV, Newman SA, Cabello FC. Borrelia burgdorferi binds to, invades, and colonizes native type I collagen lattices. Infect Immun. 2004; 72(6):3138-3146.
- Barthold SW, de Souza MS, Janotka JL, Smith AL, Persing DH. Chronic Lyme borreliosis in the laboratory mouse. Am J Pathol. 1993; 143(3):959-971.
- Cabello FC, Godfrey HP, Newman SA. Hidden in plain sight: Borrelia burgdorferi and the extracellular matrix. Trend Microbiol. 2005; 15(8):350-354.
- Costerton JW, Geesey GG, Cheng GK. How bacteria stick. Sci Am. 1978; 238:86-95.
- Donlan RM und Costerton JW. Biofilms: survival mechanisms of clinically relevant organisms. Clin Microbiol Rev. 2002; 15(2):167-193.
- Costerton JW, Cheng KJ, Geesey GG, Ladd TI, Nickel JC, Dasgupta M, Marrie TJ. Bacterial biofilms in nature and disease. Ann Rev Microbiol. 1987; 41:435-464.
- Costerton JW, Lewandowski Z, Caldwell DE, Korber DR, Lappin-Scott HM. Microbial biofilms. Ann Rev Microbiol. 1995; 49:711-745.
- Costerton JW, Stewart PS, Greenberg EP. Bacterial biofilms: a common cause of persistent infections. Science. 1999; 284(5418):1318-1322.
- Sapi E, Bastian S, Mpoy CM, Scott S, Rattelle A, Pabbati N, Poruri A, Burugu D, Theophilus PA, Pham TV, Datar A, Dhaliwal NK, McDonald A, Rossi MJ, Sinha SK, Luecke DF. Characterization of biofilm formation by Borrelia burgdorferi in vitro. PLoS One. 2012; 7(10):e48277.
- Whitchurch CB, Tolker-Nielsen T, Ragas PC, Mattick JS. Extracellular DNA required for bacterial biofilm formation. Science. 2002; 295(5559):1487.
- Alitalo A, Meri T, Comstedt P, Jeffery L, Tornberg J, Strandin T, Lankinen H, Bergström S, Cinco M, Vuppala SR, Akins DR, Meri S Expression of complement factor H binding immunoevasion proteins in Borrelia garinii isolated from patients with neuroborreliosis. Eur J Immunol. 2005; 35(10):3043-3053.
- Madsen JS, Burmølle M, Hansen LH, Sørensen SJ. The interconnection between biofilm formation and horizontal gene transfer. FEMS Immunol Med Microbiol. 2012; 65(2):183-195.
- Miklossy J, Kasas S, Zurn AD, McCall S, Yu S, McGeer PL. Persisting atypical and cystic forms of Borrelia burgdorferi and local inflammation in Lyme borreliosis. J Neuroinflammation. 2008; 5:40.
- Mursic VP, Wanner G, Reinhardt S, Wilske B, Busch U, Marget W. Formation and cultivation of Borrelia burgdorferi spheroplast-L-form variants. Infection. 1996; 24(3):218-226.
- Miklossy J, Kis A, Radenovic A, Miller L, Forro L, Martins R, Reiss K, Darbinian N, Darekar P, Mihaly L, Khalili K. Beta-amyloid deposition and Alzheimer’s type changes induced by Borrelia spirochetes. Neurobiol Aging. 2006; 27(2):228-236.
- Miklossy J, Khalili K, Gern L, Ericson RL, Darekar P, Bolle L, Hurlimann J, Paster BJ. Borrelia burgdorferi persists in the brain in chronic Lyme neuroborreliosis and may be associated with Alzheimer disease. J Alzheimer’s dis. 2004; 6:639-649.
- Sapi E, Kaur N, Anyanwu S, Luecke DF, Datar A, Patel S, Rossi M, Stricker RB. Evaluation of in-vitro antibiotic susceptibility of different morphological forms of Borrelia burgdorferi. Infect Drug Resist. 2011; 4:97-113.
- Asbrink E und Hovmark A. Classification, geographic variations, and epidemiology of Lyme borreliosis. Clin Dermatol. 1993; 11:353-357.
- Kraiczy P. Natürliche Komplementresistenz und humorale Immunabwehr bei Borrelia burgdorferi, dem Erreger der Lyme-Borreliose. Herausgeber: Prof. Dr. med. V. Brade, Shaker Verlag. 2004.
- Breitner-Ruddock S, Würzner R, Schulze J, Brade V. Heterogeneity in the complement-dependent bacteriolysis within the species of Borrelia burgdorferi. Med Microbiol Immunol. 1997; 185:253-260.
- Zipfel PF und Skerka C. Complement actor H and related proteins: an expanding family of complement-regulatory proteins? Immunol Today. 1994; 15:121-126.
- Zipfel PF und Skerka C. FHL-1/reconectin: a human complement and immune regulator with cell-adhesive function. Immunol Today. 1999; 20:135-140.
- Zipfel PF, Jokiranta ST, Hellwage J, Koistinen V, Meri S. The factor H protein family. Immunopharmacology. 1999; 42:53-60.
- Pangburn MK. Host recognition and target differentiation by factor H, a regulator of the alternative pathway of complement. Immunpharmacol. 2000; 49:149-157.
- Mason LMK, Veerman CC, Geijtenbeek TBH, Hovius JWR. Ménage á trois: Borrelia, dendritic cells, and tick saliva interactions. Trends in Parasitology. 2013; 1251:1-9.
- Kraiczy P, Skerka C, Kirschfink M, Brade V, Zipfel PF. Immune evasion of Borrelia burgdorferi by acquisition of human complement regulators FHL-1/reconectin and factor H. Eur J Immunol. 2001a; 31:1674-1684.
- Kraiczy P, Skerka C, Zipfel PF, Brade V. Further characterization of complement regulator-acquiring surface proteins of Borrelia burgdorferi. Infect Immun. 2001c; 69:7800-7809.
- Kraiczy P, Skerka C, Zipfel PF, Brade V. Complement regulator-acquiring surface proteins of Borrelia burgdorferi: A new protein family involved in complement resistance. Wien Klein Wochenschr. 2002b; 114:568-573.
- Hellwage J, Meri T, Heikkila A, Panellus J, Lahdenne P, Seppälä I, Meri S. The complement regulator factor H binds to the surface protein OspE of Borrelia burgdorferi. J Biol Chem. 2001; 276:8427-8435.
- Kraiczy P, Acker G, Barde V. Characteristics of the pathogen. In: Lyme Borreliosis and Tick-borne encephalitis, Eds. P. Oschmann, P. Kraiczy, J Halperin, and V. Brade, Uni-Med, Bremein. 1999; Seiten 20-27.
- Kraiczy P, Hunfeld K-P, Breitner-Ruddock S, Würzner R, Acker G, Brade v. Comparison of two laboratory methods for the determination of serum resistence in Borrelia burgdorferi isolates. Immunobiology. 2000a; 201:406-419.
- Kraiczy P, Hunfeld K-P, Peters S, Würzner R, Acker G, Wilske B, Brade V. Borreliacidal activity of early Lyme disease sera against complement-resistant Borrelia afzelii FEM1 wild-type and an OspC-laking FEM1 variant. J Med Microbiol. 2000b; 49:917-928.
- Kraiczy P, Skerka C, Kirschfink M, Brade V, Zipfel PF. Serum resistance of Borrelia burgdorferi correlates with binding of essential immune regulators of the factor H-protein family. Immunopharmacology. 2000c; 49:57.
- Kochi SK und Johnson RC. Role of immunoglobulin G in killing of Borrelia burgdorferi by the classical complement pathway. Infect Immun. 1988; 56:314-321.
- Kochi SK, Johnson RC, Dalmasso AP. Complement-mediated killing of the Lyme disease spirochete Borrelia burgdorferi. J Immunol. 1991; 146:3964-3970.
- Pachner AR, Dail D, Bai Y, Sondey M, Pak L, Narayan K, Cadavid D. Genotype determines phenotype in experimental Lyme-Borreliose. Ann Neurol. 2004; 56:361-370.
- Kraiczy P, Skerka C, Kirschfink M, Zipfel PF, Brade V. Mechanism of complement resistance of pathogenic Borrelia burgdorferi isolates. Intern Immunopharmacol. 2001b; 1:393-401.
- Pausa M, Cinco M, Rapagna F, Tedesco F. Complement-resistance strains of Borrelia burgdorferi exhibit a 90-kDa protein with antigenetic similarities with human CD49. Mol Immunol. 1999; 36:297 (Abstr).
- Haupt K, Kraiczy P, Wallich R, Brade V, Skerka C, Zipfel PF. In J Med Microbiol. 2008; 298 S1, 287-291.
- Kurtenbach K, DeMichelis S, Etti S, Schäfer SM, Sewell H-S, Brade V, Kraiczy P. Host association of Borrelia burgdorferi sensu lato – the key role of host complement. Trends Microbiol. 2002; 10:74-79.
- Wallich R, Pattathu J, Kitiratschky V, Brenner CH, Zipfel PF, Brade V, Simon MM, Kraiczy P. Identification an functional characterization of complement regulator-acquiring surface protein 1 of the Lyme disease spirochete Borrelia afzelii and Borrelia garinii. Infect Immung. 2005; 73:2351-2359.
- Pausa M, Pellis V, Cinco M, Giulianini PG, Presani G, Perticarari S, Murgia R, Tedesco F. Serum-resistant strains of Borrelia burgdorferi evade complement-mediated killing by expressing a CD59-like complement inhibitory molecule. J Immunol. 2003; 170:3214-3222.
- Satz N. Klinik der Lyme-Borreliose. 3., vollständig überarbeitete und erweiterte Auflage. Verlag Huber. 2010.
- Georgilis K, Peacocke M, Klempner MS. Fibroblasts protect the Lyme disease spirochete, Borrelia burgdorferi, from ceftriaxon in vitro. I Infect Dis. 1992; 166:440-444.
- Chary-Valckenaere I, Jaulca B. Champgnuelle J, Piemont Y, Mainard D, Pourel J. Ultrastructural demonstration of intracellular localization of Borrelia burgdorferi in Lyme arthritis. Br J Rheumatol. 1998; 37:468-469.
- Girschick HJ, Huppertz HI, Rüssmann H, Krenn V, Karch H. Intracellular persistence of Borrelia burgdorferi in human cynovial cells. Rheumatol Int. 1996; 16:125-132.
- Ma Y, Sturrock A, Weis JJ. Intracellular localization of Borrelia burgdorferi within human endothelial cells. Infect Immun. 1991; 59:671-678.
- Cabello FC, Codrey HP, Newman SA. Hidden in plain sight: Borrelia burgdorferi and the extracellular matrix. Trends in Microbiol. 2007; 15:350-354.
- Häupl T, Hahn G, Rittig M, Krause A, Schoerner C, Schönherr U, Kalden JR, Burmester GR. Persistence of Borrelia burgdorferi tissue from a patient with chronic Lyme borreliosis. Arthr Rheumatol. 1993; 36:1621-1626.
- Liang FT, Brown EL, Want T, Iozzo RV, Fikrig E. Protective niche for Borrelia burgdorferi to evade humoral immunity. Am J Pathol. 2004; 165:977-985.
- Mursic VP, Wanner G, Reinhardt S, Wilske B, Busch U, Marget W. Formation and cultivation of Borrelia burgdorferi spheroplast-L-form variants. Infection. 1996; 24(3):218-226.
- Brorson O und Brorson SH. In vitro conversion of Borrelia burgdorferi to cystic forms in spinal fluid, and transformation to mobile spirochetes by incubation in BSK-H medium. Infection. 1998; 26(3):144-150.
- Gruntar I, Malovrh T, Murgia R, Cinco M. Conversion of Borrelia garinii cystic forms to motile spirochetes in vivo. APMIS. 2001; 109:383-388.
- Alban PS, Johnson PW, Nelson DR. Serum-starvation-induced changes in protein synthesis and morphology of Borrelia burgdorferi. Microbiol. 2000; 146:119-127.
- Murgia R und Cinco M. Induction of cystic forms by different stress condition in Borrelia burgdorferi. APMIS. 2004; 112:57-62.
- Murgia R, Piazetta C, Cinco M. Cystic forms of Borrelia burgdorferi sensu lato: induction, development, and the role of RpoS. Wien Klein Wochenschr. 2002; 114:574-579.
- Kersten A, Poitschek C, Rauch S, Aberer E. Effects of Penicillin, Ceftriaxone, and Doxycycline on morphology of Borrelia burgdorferi. Antimicrob Agents Chemother. 1995; 39:1127-1133.
- Mursic V, Wanner G, Reinhard S, Wilske B, Busch U, Marget W. Formation and cultivation of Borrelia burgdorferi spheroblast-L-form variants. Infection. 1996; 24:118-226.
- Hovius JW, de Jong MA, den Dunnen J, Litjens M, Fikrig E, van der Poll T, Gringhuis SI, Geijtenbeek TB. Salp15 binding to DC-SIGN inhibits cytokine expression by impairing both nucleosome remodeling and mRNA stabilization. PLoSPatholg. 2008; 4:e31.
- Slamova M, Skallová A, Páleníková J, Kopecký J. Effect of tick saliva on immune interactions between Borrelia afzelii and murine dendritic cells. Parasite Immunol. 2011; 33:654-660.
- Skallova A, Iezzi G, Ampenberger F, Kopf M, Kopecky J. Tick saliva inhibits dendritic cell migration, maturation, and function while promoting development of Th2 responses. J Immunol. 2008; 180:6186-6192.
- Sa-Nunes A, Bafica A, Lucas DA, Conrads TP, Veenstra TD, Andersen JF, Mather TN, Ribeiro JM, Francischetti IM. Prostaglandin E2 is a major inhibitor of dendritic cell maturation and function in Ixodes scapularis saliva. J Immunol. 2007; 179:1497-1505.
- Sa-Nunes A, Bafica A, Antonelli LR, Choi EY, Francischetti IM, Andersen JF, Shi GP, Chavakis T, Ribeiro JM, Kotsyfakis M. The immunomodulatory action of sialostatin L on dendritic cells reveals ist potential to interfere with autoimmunity. J Immunol. 2009; 182:7422-7429.
- Oliveira CJ, Carvalho WA, Garcia GR, Gutierrez FR, de Miranda Santos IK, Silva JS, Ferreira BR. Tick saliva induces regulatory dendritic cells: MAP-kinases and Toll-like receptor-2 expression as potential targets. Vet Parasito. 2010; 167;288-297.
- Oliveira CJ, Sá-Nunes A, Francischetti IM, Carregaro V, Anatriello E, Silva JS, Santos IK, Ribeiro JM, Ferreira BR. Deconstructing tick saliva: non-protein molecules with potent immunomodulatory properties. J Biol Chem. 2011; 286:10960-10969.
- Cavassani KA, Aliberti JC, Dias AR, Silva JS, Ferreira BR. Tick saliva inhibits differentiations, maturation and function of murine bone-marrow-derived dendritic cells. Immunology. 2005; 114:235-245.
- Nanagara R, Duray PH, Schumacher HR Jr. Ultrastructural demonstration of spirochetal antigens in synovial fluid and synovial membrane in chronic Lyme disease: possible factors contribuiting to persistence of organisms. Hum Pathol. 1996; 27(10):1025-34.
- Eisendle K, Grabner T, Zelger B. Morphea: a manifestation of infection with Borrelia species? Br J Dermatol. 2007; 157(6):1189-98.
- Kraiczy P. Überleben im humanen Wirt: Wie sich Borrelien der Immunabwehr entziehen. Mikrobiologe. 2011; 21:186-192.
- Kurtti TJ, Munderloh UG, Johnson RC, Ahlstrand GG. Colony formation and morphology in Borrelia burgdorferi. J Clin Microbiol. 1987; 25(11):2054-8.
- Eggers CH, Kimmel BJ, Bono JL, Elias AF, Rosa P, Samuels DS. Transduction by phiBB-1, a bacteriophage of Borrelia burgdorferi. J Bacteriol. 2001; 183(16):4771-8.
- Dykhuizen DE, Brisson D, Sandigursky S, Wormser GP, Nowakowski J, Nadelman RB, Schwartz I. The propensity of different Borrelia burgdorferi sensu stricto genotypes to cause disseminated infections in humans. Am J Trop Med Hyg. 2008; 78(5):806-10.
- Elsner RA, Hastey CJ, Baumgarth N. CD4+ T cells promote antibody production but not sustained affinity maturation during Borrelia burgdorferi infection. Infect Immun. 2015; 83(1):48-56.
- Sapi E., Balasubramanian K., Maghsoudlou J.S., Socarras K.M., Timmeraju A.V., Filush K.R., Gupta K., Shalkh S., Theophilus P.A.S., Luecke D.F., McDonald A., Zelger B. Evidence of in vivo existence of borrelia biofilm in borrelial lymphocytomas. European Journal of Microbiology and Immunology, DOI: 10.1556/1886.2015.00049